measure command¶
The measure command is used for measuring geometries in molecules.
All of the options and preprocessors available from the process command
are also available.
$ ml measure --help
usage: mollib measure [-h] -i id/filename [id/filename ...] [-c filename] [-l]
[-s] [-m [MODELS [MODELS ...]]]
[-d atom atom | -a atom atom atom | -dih atom atom atom atom | -w atom atom]
[--stats] [--only-intra] [--exclude-intra]
[--only-intra-chain] [--exclude-intra-chain]
[--only-delta DELTA] [--only-bonded] [--hydrogenate]
[--rama]
arguments:
-h, --help show this help message and exit
-i id/filename [id/filename ...], --in id/filename [id/filename ...]
(required) The filename(s) or PDB identifier(s) of the
structure(s)
-c filename, --config filename
The configuration filename
-l List details on the molecule(s)
-s, --save Save fetched files to the local directory.
-m [MODELS [MODELS ...]], --models [MODELS [MODELS ...]]
The models numbers to analyze.
--hydrogenate Strip hydrogens and re-add them before analysis
measurement options:
-d atom atom, --dist atom atom
Measure distances between 2 atom selections. ex: 31.N
32.CA
-a atom atom atom, --angle atom atom atom
Measure angles between 3 atom selections. ex: 31.N
31.CA 31.C
-dih atom atom atom atom, --dihedral atom atom atom atom
Measure dihedral angles between 4 atom selections. ex:
31.N 31.CA 31.C 32.N
-w atom atom, --within atom atom
Measure all distances from atom selection to within
the specified distance. ex: 31:33.N 5
--stats Report statistics on the reported measurements.
--rama Report the Ramachandran angles. Filters and options
are ignored.
filters:
--only-intra Only report measurements within a residue
--exclude-intra Exclude measurements within a residue
--only-intra-chain Only report measurements within a chain
--exclude-intra-chain
Exclude measurements within a chain
--only-delta DELTA Only report residues separated by DELTA residue
numbers
--only-bonded Only report measurements from bonded atoms
Arguments¶
-d/--distatomatomMeasure the distance (in Angstroms) between two atoms.
Multiple atom pairs can used. ex:
-d 31.N 31.CA -d 32.N 33.CAAtoms must follow the standard naming conventions. See Abbreviated Selectors and Filters.
-a/--angleMeasure the angle (in degrees) between three atoms.
Multiple atom triplets can be used. ex:
-a 31.N 31.CA 31.CB -a 32.N 32.CA 32.CBAtoms must follow the standard naming conventions. See Abbreviated Selectors and Filters.
-dih/--dihedralMeasure the dihedral angle (in degrees) between four atoms.
Multiple atom quartets can be used. ex:
-dih 30.C 31.N 31.CA 31.C -dih 31.N 31.CA 31.C 32.NAtoms must follow the standard naming conventions. See Abbreviated Selectors and Filters.
Note
If simple Ramachandran dihedrals are needed, checkout
--rama.--stats- Report the average and standard deviation of all measured values. This option only applies to the distance, angle and dihedral measurements.
--ramaMeasure Ramachandran angles (in degrees) for a protein. Filters and options are ignored. Heteroatom chains are skipped.
The
--ramacommand classifies Ramachandran angles based on backbone-backbone amide hydrogen bonds.
Atom Selectors and Filters¶
Abbreviated Selectors¶
The measure methods find atoms using atom locators. Atom locators must
follow one of these conventions:
- (residue number).(atom name). ex:
31.CBfor theCBatom of residue number 31.- (chain id).(residue number).(atom name). ex:
A.31.CBfor theCBatom of residue number 31 in chain ‘A’.
Additionally, the chain id, residue number or both can be expressed as a range using the ‘:’ character:
- (residue range).(atom name). ex:
31:34.CBfor theCBatom of residue number 31, 32, 33 and 34.- (chain range).(residue number).(atom name). ex:
A:C.34.CBfor theCBatom of residue number 34 for chains ‘A’, ‘B’, ‘C’ and ‘D’.
Finally, heteroatom chains have an asterisk appended to them. ex: ‘C*’
Note
Atom selections may encompass hundreds of atoms, which when used
in combination, could lead to searches over millions of
combinations. To help improve their performance, you can either
narrow their scope by reducing the range of chains or residue
numbers, combine multiple Filters or use one of
the shortcut selectors, like --rama for Ramachandran
dihedral angles.
Filters¶
--only-intra- Exclude atom selections that are not within the same residue.
--exclude-intra- Exclude atom selections that are within the same residue.
--only-intra-chain- Exclude atom selections that are not within the same chain.
--exclude-intra-chain- Exclude atom selections that are within the same chain.
--only-deltaDELTA- Exclude atom selections that don’t have at least one set of atoms
with residues separated by
DELTAnumber. This filter ignores the chain identifier and may need to be combined with--filter-intra-chainor--exclude-intra-chain. --only-bondedExclude atom selections that are not bonded. The bonded tests linear bonding relationships. For example, a dihedral with four atoms (atom1, atom2, atom3 and atom4) must have bonds between atom1–atom2, atom2–atom3 and atom3–atom4.
Note
Bonded searches have to investigate the topology of each atom selection, which can be slower than the above filters. Combining the
--only-bondedfilter with other filters, like--only-delta 1, can significantly speed up searches.
Examples¶
Measuring distances¶
Measure \(\alpha\)-helical HA-H distances in chain ‘A’ for residues 23-49 of 2MUV, the homotetrametic influenza M2 channel. Include statistics on the measured distances.
$ ml measure -i 2MUV -d 23:49.HA 23:49.H --only-delta 3 --stats
Table: Distances for 2MUV-1
Num Atom 1 Atom 2 Dist. (A)
---- --------- -------- -----------
1 A.S23.HA A.L26.H 7.01
2 A.D24.HA A.V27.H 5.08
3 A.P25.HA A.V28.H 3.38
4 A.L26.HA A.A29.H 3.58
5 A.V27.HA A.A30.H 3.64
6 A.V28.HA A.N31.H 3.58
7 A.A29.HA A.I32.H 3.42
8 A.A30.HA A.I33.H 3.59
9 A.N31.HA A.G34.H 3.53
10 A.I32.HA A.I35.H 3.75
11 A.I33.HA A.L36.H 3.42
12 A.I35.HA A.L38.H 3.36
13 A.L36.HA A.I39.H 3.54
14 A.H37.HA A.L40.H 3.49
15 A.L38.HA A.W41.H 3.42
16 A.I39.HA A.I42.H 3.77
17 A.L40.HA A.L43.H 3.63
18 A.W41.HA A.D44.H 3.46
19 A.I42.HA A.R45.H 3.61
20 A.L43.HA A.L46.H 3.36
21 A.D44.HA A.F47.H 3.58
22 A.R45.HA A.F48.H 3.67
23 A.L46.HA A.K49.H 6.94
---------
4.0 ± 1.0
Measure CA-CA distances between residue 20-21 for chains ‘A’, ‘B’, ‘C’ and ‘D’ of 2MUV–excluding same residue distances and same chain distances.
$ ml measure -i 2MUV -d A:D.20:21.CA A:D.20:21.CA --exclude-intra --exclude-intra-chain
Table: Distances for 2MUV-1
Num Atom 1 Atom 2 Dist. (A)
---- --------- --------- -----------
1 A.N20.CA B.N20.CA 18.05
2 A.N20.CA B.D21.CA 17.50
3 A.D21.CA B.N20.CA 14.39
4 A.D21.CA B.D21.CA 13.70
5 A.N20.CA C.N20.CA 25.10
6 A.N20.CA C.D21.CA 21.94
7 A.D21.CA C.N20.CA 21.95
8 A.D21.CA C.D21.CA 19.02
9 A.N20.CA D.N20.CA 17.86
10 A.N20.CA D.D21.CA 14.21
11 A.D21.CA D.N20.CA 17.32
12 A.D21.CA D.D21.CA 13.52
13 B.N20.CA C.N20.CA 17.89
14 B.N20.CA C.D21.CA 17.44
15 B.D21.CA C.N20.CA 14.21
16 B.D21.CA C.D21.CA 13.64
17 B.N20.CA D.N20.CA 25.32
18 B.N20.CA D.D21.CA 22.15
19 B.D21.CA D.N20.CA 22.14
20 B.D21.CA D.D21.CA 19.21
21 C.N20.CA D.N20.CA 17.51
22 C.N20.CA D.D21.CA 17.00
23 C.D21.CA D.N20.CA 13.86
24 C.D21.CA D.D21.CA 13.20
Compare the distance between the HA of residue 5 and the H of residue 21 for two different structures, 2KXA and 2LWA. The 2KXA structure represents the wildtype hemagglutinin fusion peptide (HAfp) in the closed helical-hairpin structure, placing these two atoms in close promixity. The 2LWA structure represents the conformational ensemble of the HAfp-G8A mutant with a closed structure (chain ‘A’), a semi-closed structure (chain ‘B’) and an open structure (chain ‘C’).
$ ml measure -i 2KXA 2LWA -d A:C.5.HA A:C.21.H --only-intra-chain
Table: Distances for 2KXA-1
Num Atom 1 Atom 2 Dist. (A)
---- -------- -------- -----------
1 A.A5.HA A.W21.H 3.30
Table: Distances for 2LWA-1
Num Atom 1 Atom 2 Dist. (A)
---- -------- -------- -----------
1 A.A5.HA A.W21.H 3.22
2 B.A5.HA B.W21.H 11.77
3 C.A5.HA C.W21.H 18.47
Measuring Angles¶
Measure the angle of the bonded ‘C-1’–‘N’–‘H’ atoms for residues 20-30 from the ubiquitin structure 2MJB.
$ ml measure -i 2MJB -a 20:30.C 20:30.N 20:30.H --only-bonded
Table: Angles for 2MJB-1
Num Atom 1 Atom 2 Atom 3 Angle (deg)
---- -------- -------- -------- -------------
1 A.S20.C A.D21.N A.D21.H 120.2
2 A.D21.C A.T22.N A.T22.H 118.4
3 A.T22.C A.I23.N A.I23.H 119.0
4 A.I23.C A.E24.N A.E24.H 118.7
5 A.E24.C A.N25.N A.N25.H 119.2
6 A.N25.C A.V26.N A.V26.H 118.9
7 A.V26.C A.K27.N A.K27.H 119.2
8 A.K27.C A.A28.N A.A28.H 118.5
9 A.A28.C A.K29.N A.K29.H 118.4
10 A.K29.C A.I30.N A.I30.H 118.9
Measuring Dihedrals¶
The following example measures the \(\phi\) angle for residues 2-6 of the hemagglutinin fusion peptide domain (2KXA).
$ ml measure -i 2KXA -dih 2:6.C 2:6.N 2:6.CA 2:6.C --only-bonded --stats
Table: Dihedrals for 2KXA-1
Num Atom 1 Atom 2 Atom 3 Atom 4 Dihedral (deg)
---- ------- ------- -------- ------- ----------------
1 A.L2.C A.F3.N A.F3.CA A.F3.C -65.2
2 A.F3.C A.G4.N A.G4.CA A.G4.C -57.2
3 A.G4.C A.A5.N A.A5.CA A.A5.C -68.4
4 A.A5.C A.I6.N A.I6.CA A.I6.C -61.7
-----------
-63.0 ± 4.0
Ramachandran Angles¶
Measure the Ramachandran \(\phi\) and \(\psi\) angles for the hemagglutinin fusion peptide structure 2KXA.
$ ml measure -i 2KXA --rama
Table: Ramachandran angles for 2KXA-1
Residue Phi (deg) Psi (deg) Classification Minor E (kT) / Prob.
-------- ---------- ---------- --------------- ------- ----------------
A.G1 0.0 -179.9 0.0 / 100.0%
A.L2 -61.0 -45.6 alpha-helix N-term 1.1 / 31.9%
A.F3 -65.2 -48.3 alpha-helix 1.7 / 18.4%
A.G4 -57.2 -32.3 alpha-helix 1.9 / 15.4%
A.A5 -68.4 -45.0 alpha-helix 0.7 / 51.0%
A.I6 -61.7 -51.1 alpha-helix 2.5 / 8.1%
A.A7 -66.7 -42.8 alpha-helix 0.7 / 51.0%
A.G8 -63.4 -34.5 alpha-helix 1.4 / 23.5%
A.F9 -65.4 -44.3 alpha-helix 0.7 / 51.0%
A.I10 -65.1 -28.7 alpha-helix 3.4 / 3.4%
A.E11 -92.2 -47.2 alpha-helix 7.5 / 0.1%
A.G12 -111.9 28.0 alpha-helix C-term 5.1 / 0.6%
A.G13 44.9 -146.5 7.2 / 0.1%
A.W14 -49.7 -61.9 alpha-helix N-term 5.7 / 0.3%
A.T15 -49.6 -32.2 alpha-helix 8.4 / 0.0%
A.G16 -69.5 -37.4 alpha-helix 0.9 / 38.9%
A.M17 -58.3 -46.5 alpha-helix 0.8 / 45.5%
A.I18 -62.6 -49.1 alpha-helix 0.6 / 54.7%
A.D19 -55.5 -45.6 alpha-helix 0.8 / 45.5%
A.G20 -65.7 -34.8 alpha-helix 1.7 / 17.7%
A.W21 -62.9 -48.0 alpha-helix 0.6 / 54.7%
A.Y22 -77.6 -33.0 alpha-helix C-term 2.9 / 5.3%
A.G23 74.3 -87.9 6.7 / 0.1%
A.S24 -56.7 0.0 0.0 / 100.0%
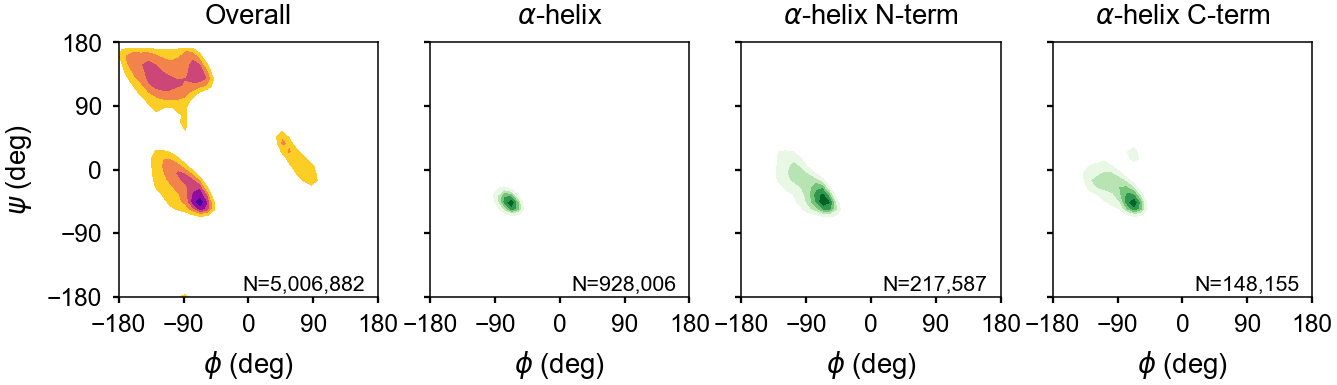
Approach to Secondary Structure Assignments¶
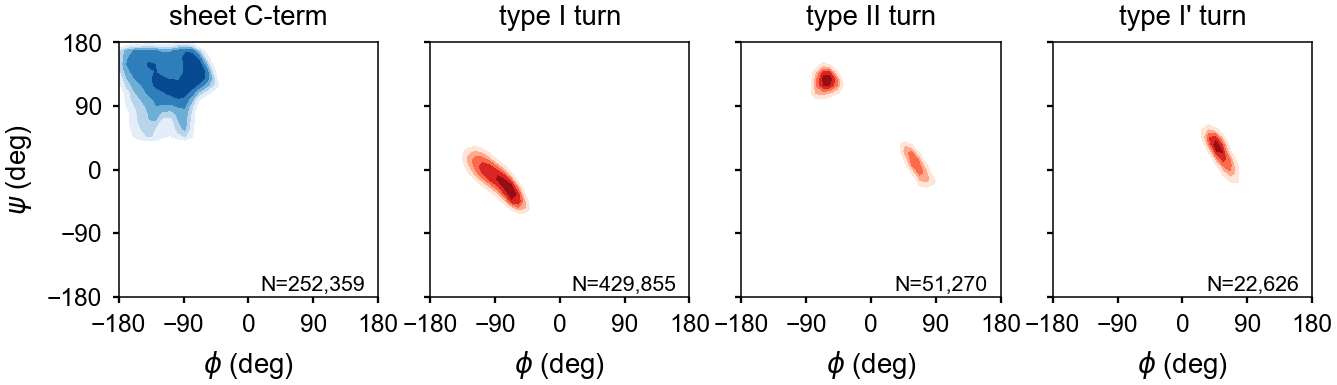
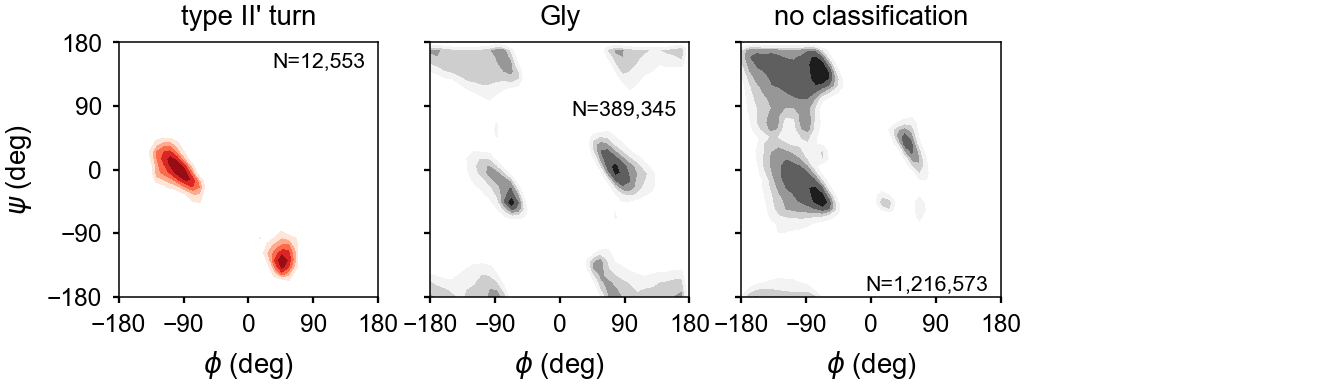
\(\beta\) Turns¶
Turns are defined by a hydrogen bond between residues ‘i’ and ‘i+4’ as well as the backbone torsion angles for residues ‘i+1’ and ‘i+2’. The turn type is based on the torsion angles of the ‘i+1’ and ‘i+2’ residues.
| Type | \(\phi_{i+1}\) | \(\psi_{i+1}\) | \(\phi_{i+2}\) | \(\psi_{i+2}\) |
|---|---|---|---|---|
| I | -60º | -30º | -90º | 0º |
| I’ | 60º | 30º | 90º | 0º |
| II | -60º | 120º | 80º | 0º |
| II’ | 60º | -120º | -80º | 0º |
Assignments of the turn residues ‘i+1’ and ‘i+2’ are made. However, since the torsion angles of the terminal residues–specifically \(\phi\) of residue ‘i’ and \(\psi\) of residue ‘i+4’–are flexible, these are not included in the assignment.
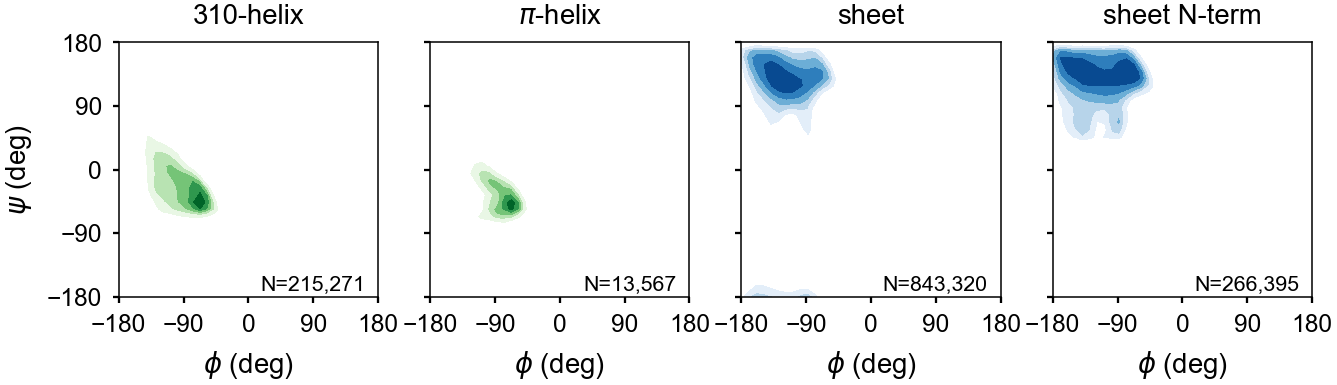
Helices¶
Helices consist of stretches of hydrogen bonded residues with helical dihedrals. 310-helices are typically short, with one or more ‘i’-‘i+3’hydrogen bonds, and these can be mischaracterized as turns (type I turns). In this case, mollib checks that all residues in the helix have helical dihedral angles.
Sheets¶
Sheets are first identified by finding hydrogen bonds between residues with sheet torsion angles. This process identifies most sheet residues. However, for strands on the edges of sheets, every second amino acid may not form an internal hydrogen bond.
To accurately identify sheet strands, mollib will find groups of sheet hydrogen bonds, then it will evaluate whether the residues are in a checkered pattern and whether the previous or subsequent residues have sheet backbone torsion angles. Thereafter, it will assign all residues in the group to a sheet classification, if no other classification has already been made. See assign blocks for details.
Assign Blocks¶
Secondary structure assignments are made based on hydrogen bonds. In some cases, such as the edge strands of sheets or short 310-helices, residues within a contiguous block are not assigned because they do not form an internal hydrogen bond. Mollib assign contiguous blocks of residues with the same secondary structure by testing the dihedral of residues within that block and filling gaps in assignment.
For example, a checkered sheet assignment (‘E E E E E’) will be assigned as a single contiguous ß-strand (‘EEEEEEEEE’) if all residues in the block have ß-strand backbone dihedral angles. 310-helices are another example in which the ‘i’ and ‘i+3’ residues are hydrogen bonded, yet the ‘i+1’ and ‘i+2’ residues may not be. In this case, the gap will be filled by assigning residues ‘i’ through ‘i+3’ as 310-helix, if all four residues have helical dihedral angles.
Additionally, assigning blocks will label the minor classification of N- and C-terminal residues for certain secondary structure blocks, depending on the settings.
Note
The identification of the ‘N-term’ and ‘C-term’ minor classifications are done separately for residues and hydrogen bonds. These assignments may be different between residues and hydrogen bonds.
Energy Maps¶
The backbone dihedral probabilities and energies are calculated from potential of mean force plots for each type of secondary structure. It is calculated from the probability of finding a particular set of dihedral angles in a group of high-resolution structures. A high probability indicates that the measured dihedral angles are observed frequently in high-resolution structures. Conversely, a low probability indicates that a particular set of dihedral angles is rarely seen in high-resolution structures. These are typically colored in yellow (relatively rare) or red (very rare).
The energies represent a potential of mean force (PMF) calculated from a Boltzmann inversion.
The energy is zero when a set of dihedrals angles is optimal for a given type of secondary structure classification. The following are the energy plots for each secondary structure classification.